

La enfermedad de Paget (EP) es una enfermedad del tejido óseo, al que afecta de forma focal, caracterizándose por una anomalía de la remodelación ósea que es excesiva y anárquica.
La EP es, junto a la osteoporosis, la osteopatía más frecuente en los países de nuestro entorno. Es singular la presencia de osteoclastos gigantes con un número de núcleos superior al habitual y el aspecto de rompecabezas del tejido óseo neoformado. La enfermedad tiene predilección por los varones, de forma discreta, aumentando la prevalencia con la edad y alcanzando un pico máximo en torno a los 65 años. Muchos pacientes no requieren tratamiento porque la enfermedad es localizada y asintomática. La presencia de inclusiones virales en las células osteoclásticas de estos pacientes puede indicar una etiopatogenia infecciosa en una edad temprana y en sujetos predispuestos genéticamente.
Epidemiología
Afecta, básicamente, a individuos mayores de 60 años siendo excepcional antes de los 40. En algunas series, el 67 por ciento de los casos oscilaba entre los 60 y 80 años. En estudios autópsicos, la prevalencia de la enfermedad en la población se halla entre el 3 y el 5 por ciento, aumentando cuando el estudio se limita a individuos de edad avanzada y existiendo entonces un leve predominio en las mujeres. La distribución geográfica mundial es irregular y hay zonas en las que es excepcional. En Inglaterra es frecuente, acumulándose los diagnósticos en determinadas zonas. Existe una fuerte tendencia a la agregación familiar, compatible con una herencia autosómica dominante o con un mecanismo multifactorial (genético y ambiental). La presencia de inclusiones parecidas a virus (paramixovirus como el sarampión, virus respiratorio sincitial o del moquillo canino) en el citoplasma y el núcleo de los osteoclastos, con positividades de los anticuerpos monoclonales dirigidos contra antígenos de los paramixovirus, hacen altamente sugestiva su relación con una infección ósea por virus. La distribución universal de los paramixovirus y la relativa localización de la enfermedad hacen pensar en una predisposición personal a desarrollarla, que no ha sido confirmada por los estudios del sistema HLA.
Para estimar los cambios asociados con la edad y la prevalencia de la EP en Gran Bretaña, los autores del artículo han realizado un análisis radiológico de la enfermedad en 10 centros sanitarios británicos de distintas ciudades, utilizando radiografías y métodos radiológicos idénticos a los utilizados en un estudio previo de 1974. De los archivos del periodo 1993-1995, se obtuvieron radiografías abdominales de la pelvis, sacro, cabezas femorales y vértebras lumbares de los sujetos mayores de 55 años. Las radiografías (9828, 4625 hombres, 5203 mujeres) de las 10 ciudades fueron evaluadas por el radiólogo que participó en el estudio de 1974. Así se obtuvo una prevalencia aproximada de la EP de un 2 por ciento con un predominio masculino (1.6 varones por cada mujer), diferencia que aumentó con la edad (6.9 y 5.8 por ciento entre hombres y mujeres mayores de 85 años, respectivamente). En 1994, la presencia de la EP en las 10 ciudades era sólo el 40 por ciento de los casos observados durante el estudio de 1974. El declive en la prevalencia estaba claro en todos los centros, pero era más marcado en aquéllos centros con prevalencias altas en el estudio original. Los autores sugieren que este estudio de la EP en 10 ciudades británicas arroja una prevalencia ajustada a la edad de 2.5 por ciento entre los hombres y 1.6 por ciento entre las mujeres mayores de 55 años, con un gran decremento en la tasa entre 1974 y 1994. Este declive hace pensar en una contribución medioambiental a la etiología de esta enfermedad que, consideran, requiere una investigación ulterior.
Fisiopatología y Anatomía Patológica
Una característica de esta enfermedad es el aumento de la resorción ósea seguido de un incremento compensatorio de la síntesis (el recambio óseo puede llegar a ser 20 veces mayor de lo normal). En una fase inicial, puede predominar la resorción (fase osteoporótica, destructiva u osteolítica), seguida de una fase mixta en la que la formación ósea se acopla a la destrucción. A medida que disminuye la actividad osteoclástica aumenta la formación de hueso duro, denso y menos vascularizado (fase osteoblástica o esclerótica).
Se considera que la principal alteración fisiopatológica radica en el aumento de la generación y en la hiperactividad de los osteoclastos. El aumento del recambio óseo depende de la extensión de la enfermedad y se relaciona con el aumento de las concentraciones plasmáticas de fosfatasa alcalina ósea.
El estudio anatomopatológico del hueso pagético muestra, como hallazgo macroscópico característico, la presencia de lesiones líticas y/o blásticas (osteosclerosis) que se pueden acompañar de deformación e hipertrofia localizadas. El estudio microscópico de las lesiones líticas pone de manifiesto un aumento típico en el número y tamaño de los osteoclastos, los cuales suelen contener más de diez núcleos. Los osteoblastos son más numerosos y de mayor tamaño. En el examen con microscopio electrónico se pueden observar en los osteoclastos inclusiones citoplasmáticas y nucleares de tipo tubular (microcilindros) que recuerdan las que aparecen en células infectadas por paramixovirus u otros corpúspulos virales. Cuando se estudia el hueso de las lesiones blásticas se observa un tejido desestructurado con líneas de cemento entrecortadas que recuerdan un rompecabezas. El estudio histológico está justificado en caso de duda diagnóstica.
La etiología de la EP ósea todavía es desconocida, aunque varios autores apuntan hacia una etiopatogenia viral. Con microscopía electrónica se han hallado características inclusiones nucleares y citoplasmáticas en los osteoclastos de los enfermos pagéticos que muestran una gran similitud con las nucleocápsides de los paramixovirus. Se observa degeneración sarcomatosa en el 2 por ciento de los pacientes afectos por la EP. La región del organizador nuclear es la parte del nucleolo involucrada en la síntesis de ribosomas, conteniendo proteínas no-histonas que pueden teñirse con plata (AgNORs) durante la interfase nuclear. El número de AgNOR se correlaciona con la actividad proliferativa de las poblaciones celulares, tanto normales como malignas. Las células cancerígenas tienen un aumento de la demanda de RNA ribosómico, encontrándose correlaciones entre la producción de RNA ribosómico y entre los antígenos AgNORs y los antígenos proliferativos. Los autores del artículo han adaptado el método de tinción para las biopsias de tejidos óseos de pagéticos sin decalcificar, para la microscopía óptica y para la microscopía electrónica. Se realizaron secciones óseas de diez enfermos de Paget, sin tratamiento previo con bifosfonatos. También se realizaron estudios similares en diez pacientes afectos por enfermedades de características patológicas similares con aumento del número de osteoclastos, como el hiperparatiroidismo secundario y primario, siendo utilizados como controles. El AgNORs aparecía como puntos negros dentro del nucleolo de los núcleos de los osteoclastos y se numeraron fácilmente. Un máximo de dos o tres puntos podía verse en los núcleos de los ostoeoclastos controles. En los osteoclastos pagéticos, el número de AgNOR fue aumentado significativamente (6.80 +/- 2.57 puntos vs 2.12 +/- 1.07 en los controles). Las inclusiones virales dentro de los núcleos aparecían teñidas débilmente y no se podían confundir con AgNORs. El número elevado de AgNORs en los núcleos de los osteoclastos pagéticos refleja una abundancia mayor de ribosomas. Como el osteoclasto pagético es muy activo, la síntesis citoplasmática de proteínas aumenta al máximo. Un aumento en AgNORs no refleja la actividad proliferativa de la célula porque los osteoclastos surgen por la fusión de sus precursores. Los autores del artículo postulan que (a) otro mRNA (de origen del viral/oncogen) podría transcribirse activamente en los pacientes afectados por la EP requiriendo más ribosomas; o (b) un genoma/oncogen viral promueve alteración del mecanismo nuclear/nucleolar.
Cuadro clínico
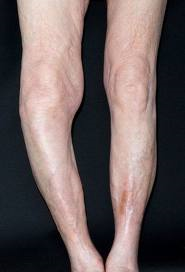
Actualmente, el diagnóstico de EP es casual en más del 60 por ciento de los casos, en pacientes que son explorados por otros motivos. En tales enfermos, la sospecha diagnóstica suele estar motivada por una elevación inexplicable de la fosfatasa alcalina y/o la observación de una imagen radiológica típica. En los casos en los que el diagnóstico se realiza por síntomas relacionados con la enfermedad, la alteración de la cadera (síndrome de cadera) es la más frecuente, seguida, en orden decreciente, por la lumbalgia, el dolor y la deformidad craneales, la cifosis progresiva, la limitación de la movilidad del hombro y las fracturas. Al ser una enfermedad que afecta a individuos mayores de 60 años, incide en un segmento de población en el que los síntomas del aparato locomotor son muy frecuentes, siendo difícil el diagnóstico diferencial. Así, el 80 por ciento de los pacientes afectos de EP experimentan síntomas no relacionables con la enfermedad y sí, en cambio, con los fenómenos degenerativos articulares o afecciones de las partes blandas. La enfermedad activa y sus consecuencias acompañan al paciente a partir del momento del diagnóstico, por lo que no son infrecuentes períodos de observación superiores a 20 años. Los individuos asintomáticos y sin signos de actividad de la afección siguen asintomáticos durante largos períodos de observación, mientras que los sintomáticos y con acusados signos de actividad (fosfatasa alcalina e hidroxiprolinuria superiores al 100 por ciento de la normalidad) son los más expuestos a la aparición de nuevos síntomas. El síndrome de cadera constituye la manifestación más relevante y se caracteriza por la aparición de dolor funcional (mecánico) y limitación progresiva de la movilidad de dicha articulación, consecuencia de la varización del cuello femoral y de las modificaciones del tamaño de la cabeza y del acetábulo. Los síntomas característicos de la enfermedad derivan de la alteración de la remodelación ósea: el dolor óseo de tipo inflamatorio, la deformidad por aumento del tamaño y de la plasticidad del hueso, el calor y los ruidos vasculares por el hiperaflujo sanguíneo del hueso afecto. Por estos motivos, los síntomas de la EP se identifican fácilmente cuando se afectan huesos de localización subcutánea, como la tibia y los del cráneo, pero no cuando los huesos comprometidos son más profundos, aumentando la dificultad diagnóstica. La localización craneofacial es frecuente y los síntomas predominantes son la sordera, la cefalea, los ruidos vasculares, la epífora y, excepcionalmente, los dolores neurálgicos. Existen cuadros neurológicos más complejos en relación con la hidrocefalia secundaria a la platibasia por hundimiento occipital, la isquemia medular en relación con fenómenos de robo vascular transitorios o compresivos por fracturas vertebrales. Otras manifestaciones clínicas son el desajuste de la articulación del hombro – en las localizaciones humerales epifisarias, que motiva una artrosis secundaria – la insuficiencia cardíaca hipercinética – infrecuente, aunque se observa en las formas muy activas y poliostóticas – y la cifosis armónica en la afección de varias vértebras dorsales. Se puede producir una elevación del gasto cardiaco. La proliferación de vasos sanguíneos, necesarios para el metabolismo óseo, produce un aumento del flujo sanguíneo en el hueso. Así, si la enfermedad afecta a más del treinta por ciento del esqueleto, se puede producir una elevación del gasto cardiaco, aunque raras veces se produce insuficiencia cardiaca.
Algunos autores afirman que las fracturas son poco frecuentes y se localizan en las vértebras dorsales y en el cuello femoral. Son posibles las seudofracturas en la convexidad de los huesos largos. Las deformidades óseas eran en el pasado la marca de la enfermedad; se producen por la tracción de grupos musculares potentes y por el crecimiento óseo. Las más características son la incurvación lateral de las tibias y del húmero, la platibasia y el crecimiento craneal. La complicación más grave de la enfermedad es la degeneración sarcomatosa, apareciendo en menos del 2 por ciento de los pacientes y es la principal causa de sarcoma óseo del adulto. La degeneración sarcomatosa se produce habitualmente en forma de osteosarcoma, aunque también es posible la aparición de unfibrosarcoma, y debe sospecharse ante la aparición de dolor muy intenso, de características inflamatorias, en un hueso afecto, junto a un incremento acelerado de los parámetros de actividad de la enfermedad, sobre todo de fosfatasa alcalina, en especial si los síntomas se localizan en el fémur, húmero, cara o pelvis. La degeneración sarcomatosa se produce siempre en el área afectada por la EP, por lo que su demostración radiológica es difícil. Se ha observado que el tumor de células gigantes tiene una significativa prevalencia en la enfermedad. Recientemente, se le han reconocido peculiaridades clínicas e histológicas de benignidad y se le denomina «tumor reparador de células gigantes». Las localizaciones epifisarias en la rodilla provocan la aparición lenta de una artropatía degenerativa por el desajuste articular y por la inestabilidad ligamentaria debida a las alteraciones de los ejes de la articulación.
La degeneración sarcomatosa es una de las complicaciones más temibles de la EP. Los autores del artículo han descrito las características radiológicas, histopatológicas y citogenéticas de una paciente de 64 años de edad a la que se le diagnosticó un condrosarcoma orbitario en el contexto de una EP. Esta mujer presentó una rápida proptosis de su ojo derecho. Se llevaron a cabo análisis con tomografía axial computerizada, exámenes histopatológicos, y análisis citogenéticos de la masa tumoral. Los datos que arrojaron los distintos cortes radiológicos de la tomografía axial computerizada de los huesos craneales frontal y temporal mostraron los cambios típicos de la EP, consistentes, sobre todo, en un aumento de la densidad ósea. Un tumor de partes blandas en la pared orbitaria lateral derecha era compatible con un sarcoma de Paget. En los estudios histológicos, mediante fluorescencia e hibridación «in situ» se confirmó el diagnóstico de condrosarcoma. Según los autores, este es el único caso documentado de condrosarcoma orbitario desarrollado en un paciente con EP. Los autores afirman que las lesiones sarcomatosas son mucho más frecuentes en otras localizaciones óseas en estos pacientes. El diagnóstico de condrosarcoma de cualquier localización debe hacernos sospechar una EP.
El dolor y las demás manifestaciones clínicas asociadas son las razones más importantes que hacen que los enfermos afectados por la EP acudan al médico. Posiblemente, la causa más frecuente de este dolor sea la osteoartritis inducida directamente, o de una forma más indirecta, por la EP. El diagnóstico diferencial con otras artropatías puede ser sumamente complicado. Muchas veces, la EP es diagnosticada por exclusión y después de un periodo sintomático de varios años. El médico de atención primaria, según los autores del artículo, debe ser capaz de establecer el origen del dolor porque el tratamiento puede ser diferente. El dolor axial o de espalda es muy común y se debe ser lo suficientemente cuidadoso en el momento del diagnóstico, antes de atribuir el dolor a la EP, ya que se pueden originar puntos de discrepancia en el resultado del tratamiento entre el médico y el paciente. Los tratamientos actuales pueden ser capaces de paliar los síntomas dolorosos originados por el crecimiento distorsionado de los huesos en la enfermedad. Las terapias modernas de las unidades del dolor pueden favorecer los resultados, haciendo el manejo de esta patología multidisciplinario.
Diagnóstico
Muchas veces se realiza por exclusión. Se basa en la demostración de una imagen radiológica característica, acompañada o no de síntomas y de la alteración de los parámetros biológicos de resorción-formación óseas. Las dificultades diagnósticas se plantean ante una degeneración sarcomatosa, en el diagnóstico diferencial de una osteoporosis circunscrita aislada y en el de las metástasis osteoblásticas, especialmente del carcinoma de próstata, enfermedad con la que coincide al afectar a un grupo de población muy similar.
Radiológico
Es característica por la coincidencia en un mismo hueso de los siguientes datos: a)áreas líticas; b) áreas blásticas, y c) aumento de tamaño óseo y deformidad. Estas alteraciones radiológicas dan al hueso afecto un aspecto basto muy característico que traduce fielmente las alteraciones histopatológicas. Habitualmente, la pelvis es el hueso que se afecta con más frecuencia, seguido del fémur, huesos del cráneo, tibia, columna lumbosacra y dorsal, costillas y clavícula. La alteración de los huesos del cráneo en la fase lítica produce una afectación característica denominada osteoporosis circunscrita, mostrando áreas de radiotransparencia nítida en los huesos frontal, parietal y occipital.
Si las lesiones craneales evolucionan adquieren un aspecto de condensación algodonosa, observándose en los huesos largos una esclerosis intensa y un patrón trabecular muy irregular. En la pelvis no es posible distinguir las fases, siendo su aspecto habitual el de esclerosis. En el síndrome de cadera se observan las lesiones pagéticas y una disminución de la interlínea articular medial, junto a esclerosis y osteofitosis, que son alteraciones propias de la artrosis secundaria. El cuerpo vertebral puede adoptar una forma característica (patrón de marco) al aumentar las estrías verticales y el refuerzo periférico, dando un aspecto «cúbico» de las vértebras. Existen formas blásticas puras denominadas «vértebras de marfil».
Un rasgo radiológico muy frecuente, básico para el diagnóstico diferencial con otros procesos, es el aumento local del tamaño óseo, secundario a la formación de hueso cortical subperióstico. Los huesos largos se arquean, el cráneo se ensancha y aumenta de grosor el diploe externo, apareciendo en la pelvis engrosamiento característico del orificio pelviano superior. Las áreas líticas producen una expansión de la cortical que puede sugerir malignidad, pudiéndose definir con la tomografía computerizada las lesiones atípicas. La gammagrafía con bifosfonatos marcados con 99mTc es de gran ayuda en la confección del mapa de la enfermedad, ya que identifica con gran precisión todas las áreas afectas. Así, con la ayuda de la gammagrafía ha descendido notablemente el porcentaje de casos monostóticos; en la serie del autor, sólo 3 de 40 eran monostóticos, lo cual representa el 7,5 por ciento del total.
Computed tomography of Paget disease of the skull versus fibrous dysplasia. Tehranzadeh J, Fung Y, Donohue M, Anavim A, Pribram HW Skeletal Radiol 1998 Dec;27(12):664-72
Para los radiólogos es, muchas veces, un desafío el análisis de los cortes que muestra la tomografía axial computerizada del cráneo sin radiografías simples previas y sin antecedentes clínicos. Las lesiones craneales de la displasia fibrosa (FD) pueden confundirse, a menudo, con las halladas en la EP. Los autores del artículo pretenden conocer las similitudes y las diferencias radiológicas de estas patologías para poder establecer un diagnóstico diferencial con la tomografía axial computerizada craneal de los pacientes que están afectados, definiendo las características radiológicas que la tomografía computerizada puede ofrecernos. Los autores del artículo examinaron con TAC el cráneo de ocho pacientes de EP y de 18 casos de displasia fibrosa (13 casos de huesos craneales y faciales, cinco casos de huesos faciales) comparándose los resultados con los obtenidos en diez casos controles. Hallaron diez signos radiológicos similares y diez signos radiológicos que mostraban diferencias entre las dos enfermedades. Los rasgos distintivos, según la frecuencia de aparición se graduaron en: (1) la alteración de la densidad, (2) la simetría, (3) la afectación de los senos paranasales, (4) el espesor de la cortical craneal, (5) la afectación del hueso esfenoides, (6) la alteración de la cavidad orbitaria, (7) la afectación de la cavidad nasal, (8) la presencia de una masa en tejido blando, (9) la afectación maxilar, y (10) la presencia de imágenes similares a las quísticas. Estos 10 signos, según los autores, mejoran la habilidad del radiólogo para diferenciar FD y PD.
Analítico
La EP no suele modificar el hemograma ni la VSG. La actividad celular de la enfermedad altera las pruebas del laboratorio relacionadas con la formación ósea, como la fracción ósea de la fosfatasa alcalina y la proteína Gla (osteocalcina), y las de resorción, hidroxiprolinuria o la FART. Las elevaciones de estos parámetros de resorción y/o formación ósea traducen la situación de la enfermedad y oscilan con ella. Así, son normales en las formas inactivas y alcanzan cifras 20 ó 30 veces superiores en fases muy activas de formas muy extensas. Tanto los datos bioquímicos de formación (fosfatasa alcalina, osteocalcina, procolágeno) como de resorción ósea (hidroxiprolina, fosfatasa ácida, piridolina y deoxipiridolina) suelen incrementarse, siendo parámetros diagnósticos orientativos de la enfermedad. De manera característica estos marcadores del recambio óseo alterado se acompañan de una calcemia y una fosforemia normales, al igual que la PTH inmunorreactiva. Algunos pacientes con la enfermedad muy activa, inicialmente, pueden presentar aumento de la calciuria y, rara vez, hipercalcemia moderada. Sólo se observan estas hipercalcemias en fases muy activas, en las degeneraciones sarcomatosas y en formas moderadamente activas en pacientes inmovilizados. La enfermedad se controla de manera objetiva mediante la observación de los parámetros mencionados. Debe recordarse que los parámetros biológicos de actividad son normales en el nueve por ciento de los pacientes, correspondientes a formas inactivas, y que casi la mitad de los casos cursa con elevaciones de los parámetros de actividad superiores al 100 por ciento de la normalidad.
Los reactantes de fase aguda, como la VSG y la proteína C reactiva, sólo se alteran en formas muy extensas y muy activas.
Novel serum markers of bone resorption: clinical assessment and comparison with established urinary indices. Woitge HW, Pecherstorfer M, Li Y, Keck AV, Horn E, Ziegler R, Seibel MJ. J Bone Miner Res 1999 May;14(5):792-801
Aunque las mediciones de los valores urinarios de los productos de la degradación del colágeno proporcionan estimaciones válidas de resorción del hueso, su aplicación clínica es complicada debido a la pronunciada variabilidad analítica y biológica. Por consiguiente, se han desarrollado inmunoensayos para la determinación de tales parámetros en suero. En este estudio, los autores del artículo evalúan la actuación de tres nuevos marcadores séricos de actividad ósea, como los telopéptidos séricos C-terminal y N-terminal del colágeno tipo I (S-CTX y S-NTX) y la sialoproteína ósea. Se compararon los resultados con las mediciones de piridinolina urinaria total, la deoxipiridinolina total, con el telopéptido C-terminal urinario de el colágeno tipo I (U-CTX) y el telopéptidos N-terminal urinario del colágeno tipo I (U-NTX). La población estudiada comprendía hombres sanos (27), mujeres premenopáusicas (30) y postmenopáusicas (31), pacientes con trastorno hepático (24), fracaso renal (30), sin cáncer de mama (24) y con cáncer mamario (30), metástasis óseas, osteoporosis vertebral primaria (27), hiperparatiroidismo primario (16), enfermedad ósea de Paget activa (18), mieloma múltiple (18), y pacientes con hipercalcemia de origen maligno antes y después del tratamiento con pamidronato (28). Los cambios en los marcadores séricos y urinarios fueron similares en la mayoría de las enfermedades óseas de origen metabólico. Sin embargo, la diferencia de los niveles de los marcadores séricos entre los controles sanos y los pacientes afectos de osteoporosis vertebral primaria, o hiperparatiroidismo primario, aumentó. En el mieloma múltiple, todos los marcadores séricos y urinarios se elevaron (p<0.05). En carcinoma de mama la afectación ósea reflejó incrementos significativos en todo los marcadores (p<0.01), exceptuando los valores de U-CTX y S-CTX. En la hipercalcemia de origen maligno, los cambios inducidos por el pamidronato en los biomarcadores eran pronunciados para U-CTX y S-CTX y S-NTX. La disfunción hepática y el fracaso renal se asociaban a niveles elevados de todos los marcadores séricos (p<0.05). Los autores afirman que las medidas séricas reflejan resorción del hueso de una forma similar que las obtenidas por mediciones de los marcadores urinarios. Puesto que los marcadores de suero superan algunas de las limitaciones de los marcadores urinarios, su uso mejora la valoración de las patologías óseas.
Tratamiento
Se han de tratar sólo los enfermos sintomáticos. Como ya se ha mencionado previamente, un gran número de pacientes no requiere tratamiento al ser la enfermedad localizada o asintomática. Se investiga sobre nuevos tratamientos en la EP, aunque el tratamiento actual se basa en el manejo de bifosfonatos y de calcitonina. Estos fármacos producen un efecto supresor sobre la actividad de la EP inhibiendo la resorción. La alteración del recambio óseo produce ciertas manifestaciones, como el dolor primario, hipercalciuria e hipercalcemia e insuficiencia cardiaca, que pueden ser tratadas con inhibidores de la resorción.
Las indicaciones del tratamiento son el dolor persistente, la compresión nerviosa, deformidad ósea de progresión rápida que dificulta la marcha, hipercalcemia e hipercalciuria, insuficiencia cardiaca, fracturas óseas de repetición y la falta de fusión de los fragmentos.
Si el paciente es sintomático y los signos de actividad son leves, los síntomas pueden estar en relación con fenómenos degenerativos articulares. En estos casos el tratamiento debe limitarse a la prescripción de analgésicos (paracetamol) y/o antiinflamatorios. Si los síntomas tienen las características propias de la EP y hay signos de actividad biológica moderada o intensa, debe iniciarse un tratamiento con fármacos moduladores de la actividad ósea. Se utilizan como moduladores óseos la calcitonina, el etidronato y la mitramicina. Si se utilizan semanas antes de la cirugía ortopédica, pueden reducir el sangrado. También responden al tratamiento supresor las fisuras corticales y las fracturas. La calcitonina de salmón por vía subcutánea, y el etidronato, tienen similar intensidad de acción si se emplean como primera elección terapéutica, aunque la calcitonina tiene superior eficacia en la supresión del dolor y en ulteriores tratamientos en enfermos ya tratados. Las dosis más elevadas de etidronato en períodos largos se acompañan de osteomalacia, por lo que su uso no es aconsejable.
En formas muy activas, y después de agotar la respuesta a dosis superiores de calcitonina o a la administración simultánea de etidronato y calcitonina, se puede ensayar la mitramicina, en perfusión intravenosa. Cuando se utiliza la mitramicina la actividad de la enfermedad decrece de forma espectacular, con mejorías clínicas importantes. Su efecto a medio plazo es menos espectacular, ya que a los 4-6 meses reaparece la actividad de la enfermedad. La duración del tratamiento, tanto con calcitonina como con etidronato, raras veces debe superar los 6 meses, puesto que ambos fármacos pierden actividad o presentan efectos adversos en períodos más prolongados. Su duración exacta es difícil de precisar y está condicionada por la mejoría de los síntomas y de los parámetros de actividad. Hoy en día y con los nuevos difosfonatos (clodronato, alendronato, pamidronato…) la enfermedad puede inactivarse durante períodos prolongados, por lo que en el futuro, el tratamiento de la enfermedad y sus indicaciones variarán sustancialmente, al poder alterar por fin, de forma sensible, su historia natural. También se está valorando la actividad que los bifosfonatos tienen en las enfermedades con resorción ósea aumentada, como diversas neoplasias osteolíticas.
A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget’s disease of bone. Paget’s Risedronate/Etidronate Study Group. Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ Am J Med 1999 May;106(5):513-20
Los autores del artículo han tratado de comparar la eficacia y tolerabilidad del risedronato oral y del etidronato en el tratamiento de la enfermedad ósea de Paget, en varios centros de los Estados Unidos. Así, 62 pacientes de 12 centros en América del Norte recibieron 30 mg diarios, durante dos meses, de risedronato y otros 61 pacientes recibieron etidronato, 400 mg diarios, durante seis meses en un estudio aleatorio, doble-ciego. Se obtuvieron los valores plasmáticos de la fosfatasa alcalina total y de la fosfatasa alcalina ósea, además del control de las cifras de deoxipiridinolina urinaria durante 12 a 18 meses.
A los 12 meses se normalizaron los valores de la fosfatasa alcalina plasmática en el 73 por ciento de los pacientes tratados con risedronato comparado con el 15 por ciento obtenido de los pacientes tratados con el etidronato (p<0.001). El tiempo medio de la normalización de los valores plasmáticos fue de 91 días en los pacientes tratados con risedronato y de más de 360 días para los pacientes tratados con etidronato (p<0.001); las recaídas fueron de un 3 por ciento en el grupo del risedronato y de un 15 por ciento en el grupo del etidronato (p<0.05). Al mes del segundo ciclo, el 18.53 por ciento de los pacientes tratados con el risedronato y el 14 por ciento del grupo del etidronato permanecían en remisión bioquímica. Los valores de la deoxipiridinolina urinarios se normalizaron en el 87 por ciento de pacientes tratados con risedronato y en el 57 por ciento de pacientes tratados con etidronato (p<0.01); los valores séricos de la fosfatasa alcalina ósea se normalizaron en el 73 por ciento de pacientes tratados con risedronato y en el 18 por ciento de los pacientes tratados con etidronato (p<0.001). Los pacientes que habían recibido etidronato previamente tuvieron una respuesta lenta al etidronato, pero no al risedronato. La disminución del dolor fue estadísticamente significativa en el grupo del risedronato, pero no en el grupo del etidronato. Se toleraron bien ambas drogas. Los autores concluyen afirmando que, aunque el tratamiento con etidronato es eficaz, el risedronato ofrece una terapia de menor duración, remisión más prolongada y favorable reducción significativa del dolor, proporcionando remisión adicional en aquellos pacientes que no respondieron de forma favorable al tratamiento previo con etidronato.
Treatment of Paget’s disease of bone with alendronate. Lombardi A Bone 1999 May;24(5 Suppl):59S-61S
Los autores del artículo afirman que el tratamiento oral con alendronato provoca una marcada supresión del recambio óseo y produce una mejoría clínica en los pacientes con EP. Los niveles de fosfatasa alcalina sérica disminuyeron intensamente a partir del tratamiento, siendo más pronunciada que la observada con el tratamiento de etidronato y calcitonina, que suelen reducir los niveles plasmáticos de la fosfatasa alcalina un promedio de un 40 a un 50 por ciento. La administración del alendronato reduce, también, notablemente los niveles urinarios de los marcadores de resorciónal tiempo que mejora radiológicamente la osteolisis pagética. La mayoría de los pacientes tratados con alendronato normalizó el nivel plasmático de fosfatasa alcalina a los seis meses de tratamiento. Es probable que la duración de la respuesta al tratamiento esté en relación con la duración de remisión. Por consiguiente, aquellos pacientes que responden al tratamiento con alendronato, sobre todo aquellos que normalizaron sus niveles de fosfatasa alcalina, probablemente mantendrán la remisión bioquímica durante varios años. De hecho, datos inéditos preliminares parecen indicar que el alendronato es capaz de producir remisión bioquímica a largo plazo en la mayoría de pacientes. Además de su eficacia, seguridad y tolerabilidad, el alendronato perfila como un fármaco muy práctico en el tratamiento de la EP, según afirman los autores.
Bisphosphonates in the treatment of malignant bone disease. Berenson JR, Lipton A Annu Rev Med 1999;50:237-48
Aquellos tumores que promueven la lisis ósea suelen tener una activación de los osteoclastos y un aumento de la resorción ósea. Los osteoclastos pueden activarse directamente por la producción de factores endógenos por un tumor maligno o por otras células no malignas. Los bifosfonatos, fármacos que se utilizan en el tratamiento de la EP, reducen la actividad osteoclástica y, de esta manera, la resorción ósea. Desde que estos agentes se mostraron efectivos en el tratamiento de otras enfermedades asociadas con el incremento de la resorción ósea, incluso la hipercalcemia y la EP relacionadas con el cáncer, diversos estudios han intentado analizar el papel que los bifosfonatos desempeñan en los pacientes con metástasis óseas osteolíticas. Los autores del artículo realizaron un estudio aleatorio, doble-ciego mostrando la eficacia de estos fármacos en la reducción de las complicaciones esqueléticas de aquellos pacientes con metástasis óseas por cáncer de mama y mieloma múltiple, aunque recomiendan una mayor profundización en esta posible nueva indicación.
Fuente: http://www.elmedicointeractivo.com/ap1/emiold/biblio/rbcn19.php